
Introduction
Hemoglobin is an essential component of blood which is mainly composed of heme and protein. Hemoglobin can malfunction or be produced in fewer amounts than required for various reasons. In males, the normal level of hemoglobin is 14-18 g/dl and in women, the range is between 12-16 g/dl (Billett H.1990). Thalassemia occurs when the production of hemoglobin is lower than normal. It is an inherited blood disorder and a type of anemia. In this article, a comprehensive review of thalassemia has been presented.
Genetic Basis
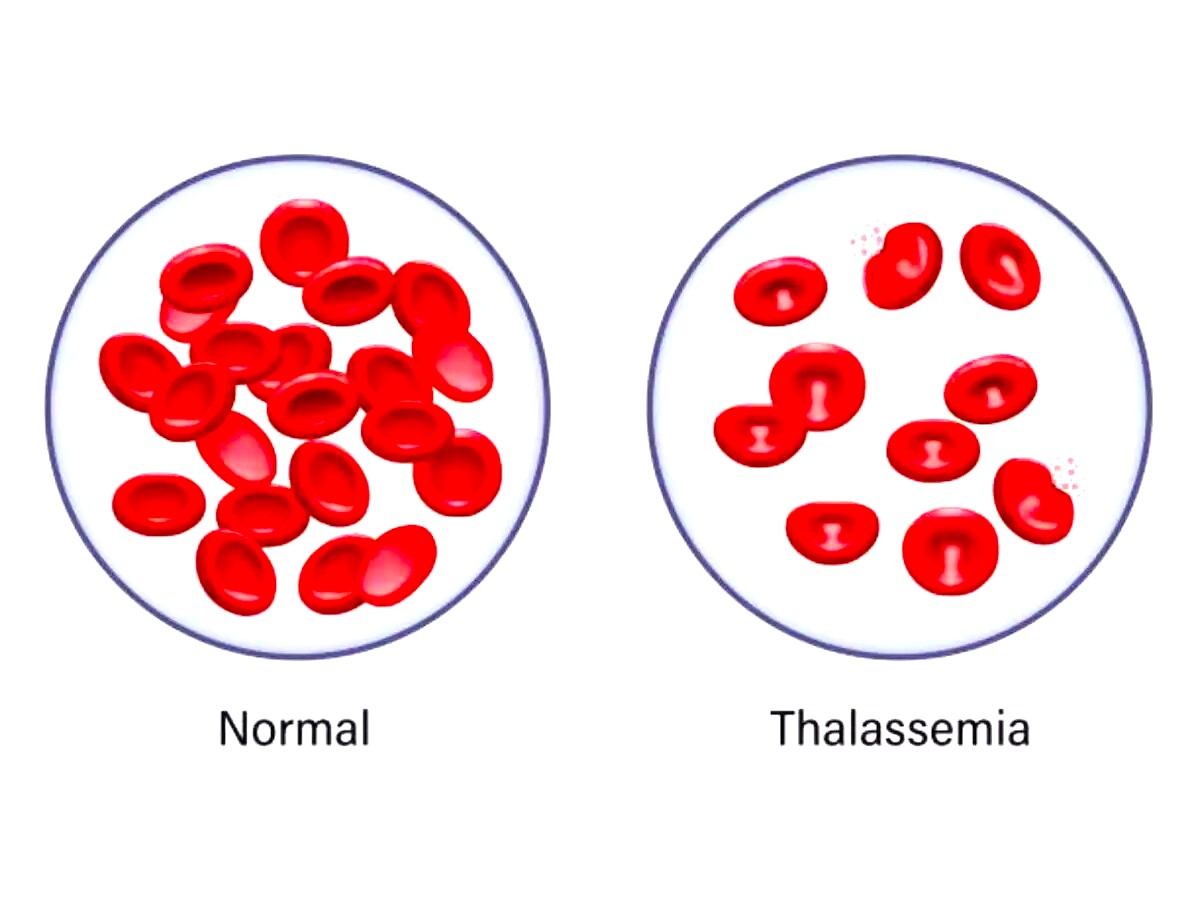
There are commonly two types of thalassemia, one is α- thalassemia and the other one is β thalassemia. α- thalassemia has two subtypes (Hb Bart’s and Hb H disease) whereas β thalassemia has three sub-types (type-1, type-2, and type-3). Thalassemia is associated with mutations. Hemoglobin has two polypeptides, namely α and β polypeptides. When α- polypeptide chain is mutated α-thalassemia arises and when β polypeptide chain is mutated β-thalassemia arises. β thalassemia is clinically serious in the homozygous state whereas α- thalassemia is lethal in the heterozygous state only in the uterus (Angastiniotis & Lobitz, 2019).
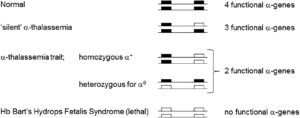
Figure 1: α- thalassemia mutation. (image derived from: https://www.sciencedirect.com/science/article/pii/S1079979617301493
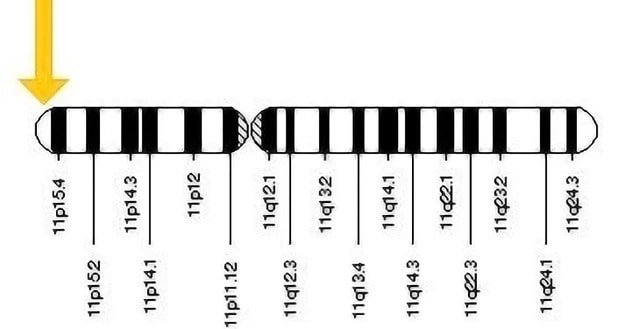
Figure 2: Mutation in 11 no chromosome is responsible for β Thalassemia. (image derived from: https://storymd.com/journal/6j4gl38unj-beta-thalassemia
α- thalassemia is mainly found in Africa, the Mediterranean Sea, and Southeast Asia populations, whereas β thalassemia is commonly found among the Middle Eastern, Asia, and Mediterranean regions. For globalization, this disorder has now spread throughout the world and commonly remains undiagnosed until anemia symptoms arrive. Asymptotic thalassemia remains undiagnosed and passed on to offspring without knowing. In this scenario, lack of awareness is making it worse.
Clinical Manifestations
According to Sheth and Thein (n/d), omissions of α-globin genes can cause an overabundance of γ-globin in the developing embryo. Deleting all four α-genes prevents HbF production during the time of 14 weeks of pregnancy, resulting in severe anemia and hydrops fetalis. When three genes are lost or altered, γ-globin (hemoglobin Barts [HbBarts]) tetramers develop first, followed by β-globin (HbH). Mutations in both β-globin genes result in reduced or nonexistent HbA production throughout the third trimester, as well as increasing anemia after birth. Characterizing complex with heterozygotes with β-thalassemia and HbE mutations yields comparable results but alongside the inclusion of HbE.
Both types of thalassemia show numerous health conditions, such as; bone deformities, dark urine, iron overload, jaundice, delayed puberty, shortness of breath, fatigue, heart problems, hepatosplenomegaly, pale skin, infection, osteoporosis, poor appetite, swollen abdomen, dizziness, growth deficiency, heart palpitations, etc. The reason behind skeletal changes is bone marrow expansion. Bone marrow is active more than usual to produce enough red blood cells in the body. Therefore, bone marrow is expanded to some extent, bore deformities are seen, and delayed puberty, growth deficiency, osteoporosis, etc. come along with it. As the RBC count falls, dizziness, paleness, and breathlessness are experienced. Moreover, as a low amount of RBC cannot send enough oxygen to tissue, for this reason, several heart diseases may surface (Karakas et al.,2016).
Diagnosis for Thalassemia
Complete Blood Count (CBC)
CBC shows results for RBC, WBC, and Platelet counts which give an idea about abnormal cell count and anemia. Thalassemia has no direct impact on WBC and platelet count. Also, CBC count does not differentiate whether the patient has α– thalassemia or β thalassemia but it can detect severity of anemia (Galanello et al.,2010).
Electrophoresis
Hemoglobin electrophoresis is a blood test where different patterns of hemoglobin are identified and it gives highly accurate results (Pang et al.,2023).
Iron studies
Iron studies check for iron deficiency or iron overload. It measures iron, ferritin, and transferrin in the blood sample. It is also highly accurate in determining thalassemia (Brancaleoni et al.,2016).
Genetic or molecular testing
Molecular testing is highly accurate and reliable. Rt-PCR, Gap-PCR, reverse dot-blot (RDB) analysis, allele-specific PCR, and direct DNA sequencing are the types of testing that can identify the mutation in the hemoglobin gene.
World Epidemiology
In a study by Musallam et al., 2023, where populations from regions of North America (which incorporates the United States and Canada), Europe (England. Denmark, Greece, Netherlands, and Spain), and Asia (specifically, Malaysia) were investigated for thalassemia, the authors found that the observed prevalence statistics varied greatly, with Spain having the lowest frequency at 0.03 per 100,000 persons between 2014 and 2017, and Malaysia having the highest rate at 4.5 per 100,000. In the USA, α–thalassemia frequency was investigated in two distinct studies. One research, which pooled information obtained from Canada and the United States from 2001 to 2004, indicated a rate of 0.04 for every one million individuals. In contrast, another research that focused primarily on the United States from 2004 to 2008 and used a strict case definition found a higher frequency of 0.6 per million individuals. These data highlight the variation in α–thalassemia prevalence between people and geographical locations. The precise statistics on the birth prevalence of thalassemia in the Middle East show rates ranging from 23.9 to 40 for every 100,000 births for beta-thalassemia major in countries like Iran and Oman. The evidence reveals the considerable burden of thalassemia in this region, emphasizing the importance of integrated public health initiatives and treatment techniques. Similarly, in Iraq, the frequency of β-thalassemia has been shown to range between 35.7 and 49.6 per one million (Musallam et al.,2023).
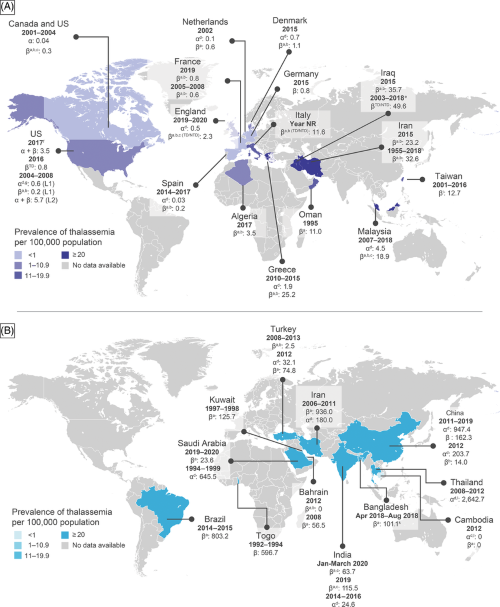
Figure 3: World prevalence of thalassemia (image derived from https://www.researchgate.net/publication/371866173_Epidemiology_of_clinically_significant_forms_of_alpha-_and_beta-thalassemia_A_global_map_of_evidence_and_gaps/citations Treatment Strate
Treatment Strategies
Blood transfusion
TDT (transfusion-dependent thalassemia) patients require time-to-time blood transfusions. Usually, every 3-4 weeks transfusions are needed for 4 hours straight each time.
Iron Chelation Therapy
Those who undergo blood transfusion suffer from iron overload. Medications are provided to eliminate the extra iron associated with blood transfusions. Iron chelation treatment medications include Deferasirox, Deferiprone, and Deferoxamine. Common negative consequences include rashes, nausea, and visual or hearing loss (Mohamed, 2017).
Blood bone marrow transplant
It is also called hematopoietic stem cell transplant (HSCT). When these stem cells do not function properly, transplant is the only way of dealing with this disease. HSCT is an effective means of treating thalassemia major as it offers patients the possibility of a cure as well as an improved standard of living (Algeri et al., 2023).
Risks Associated with Thalassemia Treatment
Complications due to iron overload in Transfusion dependent thalassemia (TDT) and Non-Transfusion dependent thalassemia (NTDT)
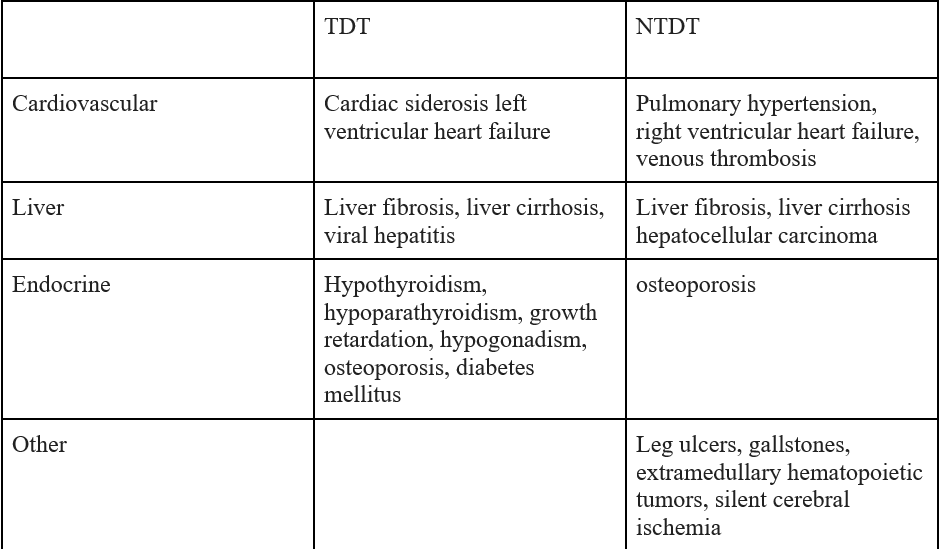
Figure 4: Complications related to iron overload in Transfusion dependent thalassemia (TDT) and Non-transfusion dependent thalassemia (NTDT)(image derived from https://ashpublications.org/hematology/article/2017/1/265/21171/Iron-overload-in-thalassemia-different-organs-at
Bone marrow Transplant challenges
Despite being a final therapeutic option, bone marrow transplant or Hematopoietic stem cell transplant (HSCT) presents problems in treating β-thalassemia major, including the necessity for eligible donors, the danger of graft-versus-host disease, and possible consequences (Granot & Storb, 2020).
Alloimmunization
Alloimmunization is one of the major threats for individuals with TDT, with 14.5% developing it. A study by Aimmatul et al., 2023 found no statistically significant relationship between transfusion frequency and alloimmunization prevalence.
Prevention and Genetic Counseling
Premarital testing is the first and foremost step for discovering and controlling thalassemia since it identifies carriers and lowers the chance of thalassemia major births. Also, prenatal testing plays a key role in the prevention and management of thalassemia. Both premarital and prenatal testing give insight into preparing for upcoming clinical procedures (Zhu et al.,2023).
Genetic counseling decreases distress in families who have thalassemia children, underlining the value of help for households dealing with this hereditary disease (Setiawan, 2017). A study found that genetic guidance for the β-thalassemia trait improved understanding, mindsets, and marital decisions while having no adverse psychological consequences (Macklipkin et. al.,1984).
Single-molecule real-time technology (SMRT) is a novel method for detecting thalassemia by finding rare variants and analyzing phenotype-genotype relationships more rapidly and precisely than existing methods (Luo et al., 2021). Also, Third-generation sequencing (TGS) technologies, like single-molecule real-time sequencing, are transforming thalassemia diagnostics and offering higher identifying rates and precision than previous methods.
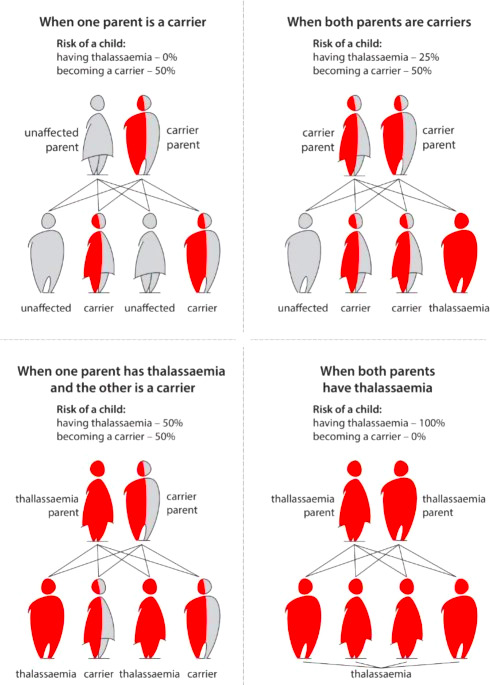
Figure 5: A comprehensive figure of pass-on Thalassemia to the off-springs. (image derived from https://www.nature.com/articles/s41415-022-5302-7
Conclusion
In this article, a comprehensive review of thalassemia has been presented. In summary, thalassemia, a congenital blood condition affecting hemoglobin synthesis, impairs the body’s capacity to transport oxygen efficiently. Timely diagnosis of this disease is crucial which involves blood testing and genetic analysis. In serious cases, therapeutic options include iron chelation therapy, blood transfusions, and lastly hematopoietic stem cell transplant. Comprehensive therapy needs continuous monitoring, patient education, and emotional support. Continued research is critical for discovering novel treatments, such as gene editing, and refining existing medications to reduce adverse effects.
References
- Aghisna Rahma Putri Wasono, Aisha Fairuz Zahira, & Hartono Kahar. (2022, December 27). Bone Marrow Transplant as Definitive Therapy for β-Thalassemia Major Patients: A literature review. https://www.ijrp.org/paper-detail/4253
- Aimmatul, Yumna, Nihayatul, Wafa., Betty, Agustina, Tambunan., Mia, Ratwita, Andarsini. (2023). The Correlation Between Frequency of Transfusion and Alloimmunization in Transfusion-Dependent Thalassemia Patients. International journal of scientific advances, doi:10.51542/ijscia.v4i1.18
- Algeri, M., Lodi, M., & Locatelli, F. (2023). Hematopoietic stem cell transplantation in thalassemia. Hematology/Oncology Clinics of North America, 37(2), 413–432. https://doi.org/10.1016/j.hoc.2022.12.009
- Angastiniotis, M., & Lobitz, S. (2019). Thalassemias: An Overview. International Journal of Neonatal Screening, 5(1), 16. https://doi.org/10.3390/ijns5010016
- Billett, H. H. (1990). Hemoglobin and hematocrit. Clinical Methods – NCBI Bookshelf. https://www.ncbi.nlm.nih.gov/books/NBK259/
- Brancaleoni, V., Di Pierro, E., Motta, I., & Cappellini, M. D. (2016). Laboratory diagnosis of thalassemia. International Journal of Laboratory Hematology, 38(S1), 32–40. https://doi.org/10.1111/ijlh.12527
- Galanello, R., & Origa, R. (2010). Beta-thalassemia. Orphanet Journal of Rare Diseases, 5(1). https://doi.org/10.1186/1750-1172-5-11
- Granot, N., & Storb, R. (2020). History of hematopoietic cell transplantation: challenges and progress. Haematologica (Roma), 105(12), 2716–2729. https://doi.org/10.3324/haematol.2019.245688
- Karakaş, S., Tellioğlu, A. M., Bilgin, M., Ömürlü, İ. K., Çalışkan, S. Ö., & Coşkun, S. (2017). Craniofacial characteristics of thalassemia major patients. The Eurasian Journal of Medicine, 48(3), 204–208. https://doi.org/10.5152/eurasianjmed.2016.150013
- Luo, S., Chen, X., Zeng, D., Tang, N., Yuan, D., Zhong, Q., Mao, A., Xu, R., & Yan, T. (2021). The value of single-molecule real-time technology in the diagnosis of rare thalassemia variants and analysis of phenotype–genotype correlation. Journal of Human Genetics, 67(4), 183–195. https://doi.org/10.1038/s10038-021-00983-1
- Mohamed, S. (2017). Thalassemia major: transplantation or transfusion and chelation. Hematology/Oncology and Stem Cell Therapy (Online), 10(4), 290–298. https://doi.org/10.1016/j.hemonc.2017.05.022
- Musallam, K. M., Lombard, L., Kistler, K., Arregui, M., Gilroy, K., Chamberlain, C., Zagadailov, E., Ruiz, K., & Taher, A. T. (2023a). Epidemiology of clinically significant forms of alpha‐ and beta‐thalassemia: A global map of evidence and gaps. American Journal of Hematology (Print), 98(9), 1436–1451. https://doi.org/10.1002/ajh.27006
- Setiawan, H., Ediati, A., & Winarni, T. I. (2017a). Genetic Counseling to Reduce the Level of Depression in Parents of Children with Thalassemia Major. 2nd International Conference on Sports Science, Health and Physical Education. https://doi.org/10.5220/0007055801020106
- Shaji, R. V., Srivastava, A., Chandy, M., & Krishnamoorthy, R. (2000). A single tube multiplex PCR method to detect the common alpha + thalassemia alleles. Blood, 95(5), 1879. https://doi.org/10.1182/blood.v95.5.1879
- Sheth S, & Thein S Thalassemia: a disorder of globin synthesis. Kaushansky K, & Prchal J.T., & Burns L.J., & Lichtman M.A., & Levi M, & Linch D.C.(Eds.), [publicationyear2] Williams Hematology, 10e. McGraw-Hill Education. https://hemonc.mhmedical.com/content.aspx?bookid=2962§ionid=252528863
- Treatment of thalassemias. (n.d.). Hematology-Oncology Associates of CNY. https://www.hoacny.com/patient-resources/blood-disorders/what-thalassemias/other-names-thalassemias/treatment-thalassemias#:~:text=Doctors%20use%20three%20standard%20treatments,re%20used%20much%20less%20often
- Pang, X., Du, H., Yang, Y., Zhou, X., Tang, N., Liu, J., & Xu, Y. (2023). [Causes of abnormal hemoglobin electrophoresis]. PubMed, 31(3), 830–836. https://doi.org/10.19746/j.cnki.issn.1009-2137.2023.03.031
- Zhan, L., Gui, C., Wei, W., Liu, J., & Gui, B. (2023). Third generation sequencing transforms the way of the screening and diagnosis of thalassemia: a mini-review. Frontiers in Pediatrics, 11. https://doi.org/10.3389/fped.2023.1199609
- Zhu, S., Yin, J., Luo, Y., Chen, Y., Lin, Z., Fu, X., Li, H., & Su, H. (2023). Clinical experience using peripheral blood parameters to analyse the mutation type of thalassemia carriers in pregnant women. Journal of Obstetrics and Gynaecology, 43(1). https://doi.org/10.1080/01443615.2023.2195490
If you like this article, you can go through our other top articles
- Vaccination: Debunking Some Common Myths – https://learnlifescience.com/vaccination-debunking-some-common-myths/
- DNA Sequencing Methods – https://learnlifescience.com/dna-sequencing-methods/
- Biosensors as a Disease Diagnostic Tool: A Comprehensive Review – https://learnlifescience.com/biosensors-as-a-disease-diagnostic-tool-a-comprehensive-review/
- An Overview of Bacterial Genomic DNA Isolation – https://learnlifescience.com/an-overview-of-bacterial-genomic-dna-isolation/
“Since the article has been written to reflect the actual views and capabilities of the author(s), they are not revised for content and only lightly edited to be confirmed with the Learn life sciences style guidelines”








2 Responses to “A Comprehensive Review of Thalassemia”