

Abstract
The demand for fast and inexpensive DNA sequencing data has given birth and dominance to a new generation of sequencing technology. So-called ‘next-generation’ sequencing technologies enable the generation of data by sequencing large amounts of DNA in parallel using a variety of methods. Although they broke new ground in genomics research, they did not recognize many of the possibilities and applications of these new technologies. Many scientists now accept that sequence analysis provides an increasingly useful approach for characterizing biological systems. They consider data sequencing and DNA sequencing to be valuable and often essential aspects of their work. Projects are already underway to map and sequence the entire genomes of organisms such as Escherichia coli, Saccharomyces cerevisiae, and Caenorhabditis. The main purpose of this review article is to discuss the different methods/technologies of DNA sequencing methods, DNA sequencing history, Key principles, and clinical applications of the next generation DNA sequencing, Next Generation Sequencing of Golden Tools in the Forensic Toolkit, principles of Next Generation sequencing, Next-generation sequencing strategy, and future possibilities.
Introduction
DNA sequencing has revolutionized the science of molecular genetics. Many scientists now accept that sequence analysis provides an increasingly useful approach for characterizing biological systems. Projects are already underway to map and sequence the entire genomes of organisms such as Escherichia coli, Saccharomyces cerevisiae, Caenorhabditis elegans, and Homo sapiens. DNA sequencing acts as a catalyst to stimulate future research in various scientific fields. Large-scale sequencing projects such as the Human Genome Project generate her DNA sequences for many unknown genes. Such data will drive the application of reverse genetics techniques to molecular biologists. Several new approaches have been developed, such as sequencing by hybridization and sequence detection using mass spectrometry and biochips. Although the average read length has increased significantly, there is still a limit to the size of target DNA that can be sequenced in a single run. (DNA Sequencing | Springer Nature Experiments, n.d.). “Massively parallel” DNA sequencing is capable of sequencing a large number of different DNA sequences in a single reaction (ie, in parallel). All next-generation sequencing (NGS) technologies monitor the addition of nucleotides to immobilized, spatially arranged DNA templates, but differ greatly in how these templates are generated and investigated. Next-generation sequencing platforms are helping to open up entirely new areas of biological research, such as studying ancient genomes and characterizing ecological diversity. (Rizzo & Buck, 2012) .
Maxam and Gilbert Method
The Sanger method is faster and easier to perform than the Maxam-Gilbert method but remains the preferred choice for most sequencing applications. A major advantage of chemical degradation sequencing is that the sequence is obtained from the original DNA molecule rather than from an enzymatic copy. This allows the analysis of DNA modifications such as methylation and the investigation of protein-DNA interactions.
The advantage of this method is that the purified DNA can be read directly. Homopolymer DNA runs are sequenced as efficiently as heterologous DNA sequencing to analyze DNA-protein interactions (e.g. footprinting) to analyze the nucleic acid structure and epigenetic modifications of DNA. (DNA Sequencing | Springer Nature Experiments, n.d.).
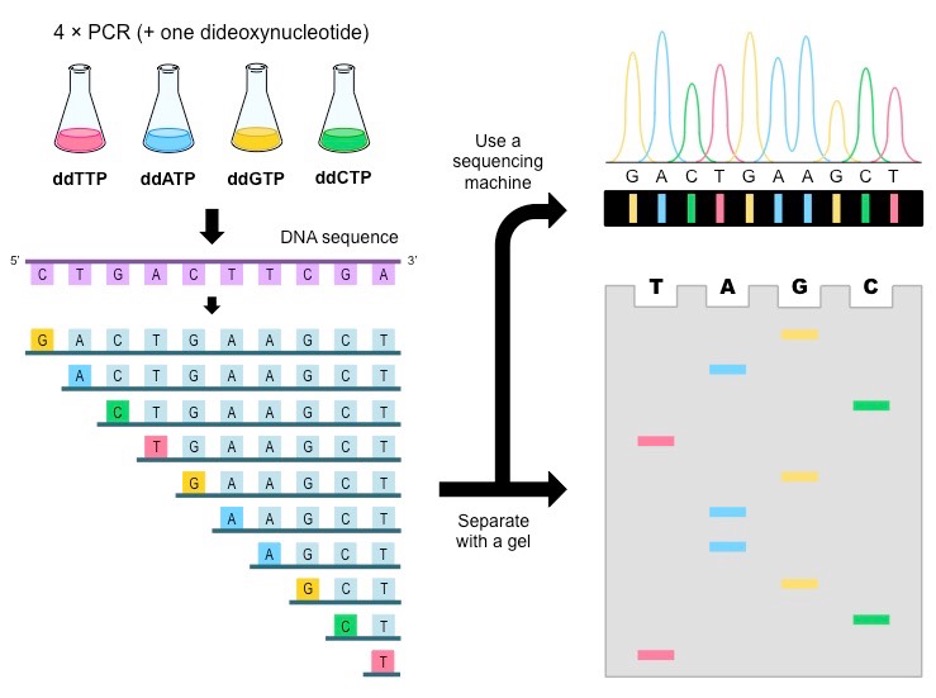
Figure 1.1: The figure shows the Maxam-Gilbert Method.
Sanger Method
The Sanger method (or chain termination) method (I) involves the synthesis of a DNA strand from a single-stranded template by a DNA polymerase. This process is based on dideoxynucleotides (ddNTPs) that are incorporated into the growing chain. By using an appropriate ratio of dNDTs in reaction with a DNA polymerase, a population of polynucleotide strands is generated. (DNA Sequencing | Springer Nature Experiments, n.d.)
In Sanger sequencing, a DNA primer complementary to the template DNA (the DNA to be sequenced) is used as the starting point for DNA synthesis. Four dideoxynucleotides triphosphates labeled with different fluorescent dyes are used to terminate the synthesis reaction. Reaction products are loaded into four lanes of a single gel and subjected to gel electrophoresis, depending on the chain-terminating nucleotides. (cdadmin, n.d.)

Figure 2.1: The figure shows the steps of the Sanger sequencing method. (cdadmin, n.d.)
Cycle Sequencing
Cycle sequencing is a new and innovative approach to dideoxy sequencing. This method uses a single primer to linearly amplify a region of template DNA. Reactions are performed 20-30 times instead of just one under the control of a thermal cycler (or PCR machine), yielding more and better sequence data with less template. (DNA Sequencing | Springer Nature Experiments, n.d.)

Figure 3.1: Scheme of the cycle sequencing reaction. Double-stranded or single-stranded templates undergo three thermocycling reactions: high-temperature denaturation, primer annealing, and primer extension/termination.
Shotgun DNA Sequencing
The shotgun sequencing method consists of several independent steps. These steps are detailed below and summarized in Figure 4.1. Briefly, DNA is sonicated to obtain fragments of the desired size. Sonicated material is end-repaired using T4 DNA polymerase and Klenow enzyme and fractionated by agarose gel electrophoresis. Size-selected fragments (usually in the 1.5-3 kb size range) are purified and inserted into a sequencing vector such as M13 by blunt-end ligation. After ligation, use the DNA to transform a suitable E. coli host. Templates are sequenced using a linear amplification method and analyzed with an automated DNA sequence. Raw data are processed to remove low-quality vector and tail sequences and assembled computationally using modern software. (Shotgun Sequencing – an Overview | ScienceDirect Topics, n.d.)
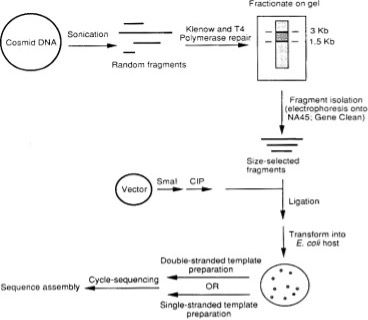 Figure 4.1: Flowchart of the shotgun method used for automated DNA sequencing. Intact cosmid DNA (or self-ligated genomic inserts) is fragmented by sonication at 4°C to an average fragment size of 2–4 kb. The sonicated DNA is end-repaired twice with a mixture of T4 DNA polymerase and Klenow enzyme and size-fractionated on an agarose gel. DNA fragments in the desired size range (1.5–3 kb) were purified by electrophoresis on NA45 paper or by Gene Clean, digested with SmaI, and dephosphorylated with calf intestinal phosphatase sequencing vector (M13, Blue script). (CIP). The ligation mixture is used to transform E. coli-competent cells. Grow white transformants, isolate double- or single-stranded DNA, and sequence using cycle sequencing protocols and an ABI 373A DNA sequencer. Sequences are assembled on a computer using modern software. (Shotgun Sequencing – an Overview | ScienceDirect Topics, n.d.)
Figure 4.1: Flowchart of the shotgun method used for automated DNA sequencing. Intact cosmid DNA (or self-ligated genomic inserts) is fragmented by sonication at 4°C to an average fragment size of 2–4 kb. The sonicated DNA is end-repaired twice with a mixture of T4 DNA polymerase and Klenow enzyme and size-fractionated on an agarose gel. DNA fragments in the desired size range (1.5–3 kb) were purified by electrophoresis on NA45 paper or by Gene Clean, digested with SmaI, and dephosphorylated with calf intestinal phosphatase sequencing vector (M13, Blue script). (CIP). The ligation mixture is used to transform E. coli-competent cells. Grow white transformants, isolate double- or single-stranded DNA, and sequence using cycle sequencing protocols and an ABI 373A DNA sequencer. Sequences are assembled on a computer using modern software. (Shotgun Sequencing – an Overview | ScienceDirect Topics, n.d.)
Polony Sequencing
Polony DNA is a non-electrophoretic sequencing method that eliminates in vitro cloning artifacts and has a lower cost per base than traditional Sanger sequencing. The supporting protocol describes how to optimize the amount of library DNA used as an ePCR template. Millions of short reads are generated in parallel via a circular DNA sequencing strategy that uses T4 DNA ligase to label each microbead with a fluorescent label. The emulsion PC protocol presented in this unit is a variation of that proposed by Dressman et al. (2003), but should not be considered the only applicable amplification method. Different sequence biochemistry can also be used. During the development of the Polony sequencing platform, many alternative polymerase- and ligase-driven approaches were evaluated. (Porreca et al., 2006)
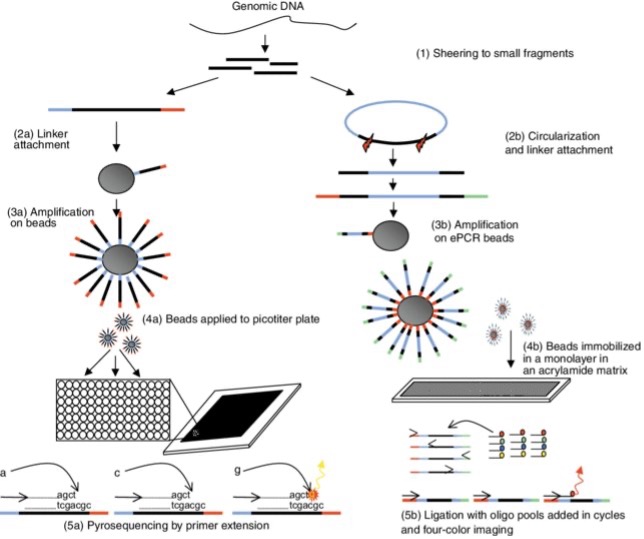
Figure 5.1: An overview of the 454 and Polony sequencing process. Both systems first fragment the genomic DNA (step 1) and then use a process of in vitro cloning followed by amplification. The 454 processes are shown on the left and the Polony sequence is on the right. In the 454 protocol, linkers are ligated to the ends of the DNA (step 2a). In polony sequencing, circularization is followed by linearization to add a linker to generate two fragments with a spacer in between and a linker at the end (Step 2B). Both processes then bind the in vitro clones to beads and run PCR on the emulsion mix to generate beads containing many clonal copies of the target fragment (steps 3a/3b). The sequencing step requires the beads to be immobilized in a monolayer so that the reaction reagents can be imaged in an environment flowing over the beads. For 454 sequencing, picotiter plates are used and contain one bead for most cells (step 4a). The Polony method immobilizes beads in an acrylamide matrix in a dense monolayer (step 4b). These methods are very similar in terms of sequencing reactions. For 454 sequencing, the DNA synthesis reaction is run from a single sequencing primer. Bases flow individually over the picotiter plate and incorporation is detected by light emission (step 5a). The Polony method uses ligation to fix primers that can be annealed at one of four positions. At each cycle, a population of fluorescently labeled degenerate 9-mers is added to the monolayer and only complementary oligos are annealed and ligated to the anchor primer. (Hall, 2007)
Next-generation sequencing (NGS): The golden tool of the forensics toolbox
DNA analysis is the cornerstone of modern forensics. Next-generation sequencing holds great potential to advance and expand molecular applications in forensics by overcoming the pitfalls of traditional sequencing methods. NGS simultaneously uses a large number of genetic markers containing high-resolution genetic data. A standard forensic technique is STR profiling. Conventional capillary electrophoresis (CE) relies on detecting the size of DNA fragments. Analyzing mixtures of DNA whose DNA comes from more than one person is a major challenge. Many studies used different sequencing systems to detect her STR loci on both autosomes and sex chromosomes in the same analysis. SNPs are single-base substitutions or insertions/deletions in the genome that account for most of the genetic variation in humans. Many features, such as skin, eye color, and hair color, are 80-90 accurate, but height predictions are less accurate. Many of the technical issues associated with STRs do not exist in SNPs, so replacing STRs with SNPs is very beneficial. Deep sequencing technology can simultaneously analyze both phenotype and identity SNPs using STRs and mitochondrial DNA (mtDNA). Recently, the entire mitochondrial genome of 25–50 individuals can be analyzed on a single Ion 316 chip (Ion Torrent), enabling inexpensive analysis of SNP polymorphisms in both regulatory and coding regions. (Aly & Bolbol, 2015)
Roche/454 FLX Pyro sequencer
The Roche/454 sequencer was first commercially introduced (2004) and uses an alternative sequencing technique known as pyrosequencing in which one nucleotide is incorporated into a library of other nucleotides. The amount of light produced is proportional to the number of incorporated nucleotides (up to detector saturation). PTPs are an integral part of the Roche/454 sequencing instrument, where each nucleotide solution is stepwise introduced into the flow cell, with each incorporation step followed by an imaging step. This strategy allows the 454 base-calling software to tune the light emitted by individual nucleotide incorporation, eliminating errors during base-calling. (Mardis, 2008).

Figure 7.1: Roche 454 GS FLX sequence. Template DNA is fragmented, end-repaired, ligated with adapters, and clonally amplified by emulsion PCR. After amplification, beads containing sequencing enzymes are placed in the wells of the picotiter plate. A picotiter plate serves as a flow cell in which iterative pyrosequencing is performed. A nucleotide incorporation event releases pyrophosphate (PP i), resulting in well-localized luminescence. APS, adenosine-5-phosphate. (Voelkerding et al., 2009).
Illumina Genome Analyzer
Illumina systems use a sequencing-by-synthesis approach. In this approach, all four nucleotides are simultaneously added to the flow cell channel along with DNA polymerase and assembled into oligo primer clustered fragments. Each nucleotide has a base-specific fluorescent label and the 3-OH group is chemically blocked, making each incorporation a unique event. A quality check pipeline evaluates the data from each run and removes sequences with poor quality. (Mardis, 2008).
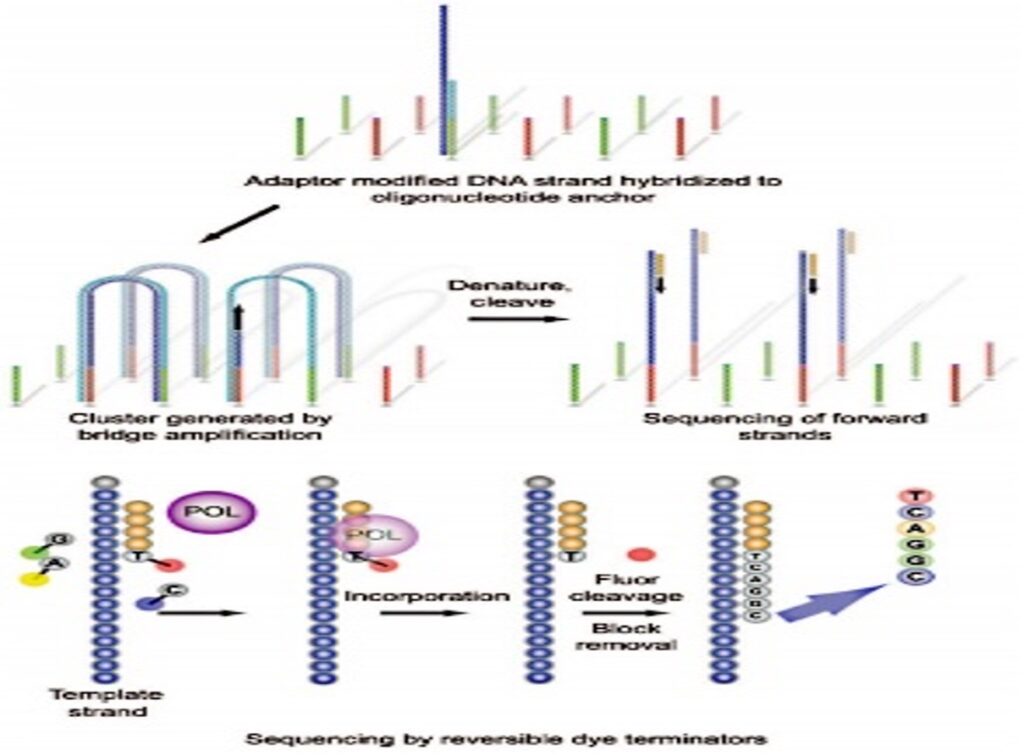
Figure 8.1: The Illumina Genome Analyzer Sequencing is the first commercially available Illumina platform. (Figure 4. Illumina Genome Analyzer Sequencing, the First of the Illumina Platforms to Be Commercially Launched., 2016)
Applied Biosystems SOLiDTM Sequencer
The SOLiD platform uses adapter ligation. Fragment libraries are similar to other next-generation platforms, using emulsions. A small magnetic bead PCR approach to amplify fragments for sequencing. Each sequencing run generates 2–4 GB of DNA sequencing data. Once the reads have been base called, have a quality score, and low-quality sequences have been removed, they are aligned to the reference genome. The massively parallel scope of next-generation sequencing implies a similarly extensive range of analysis, including image analysis, signal processing, background subtraction, and quality assessment to produce the final sequence reads for each run. Some applications are better suited for certain platforms than others, as explained below. Reading length and error profile issues require platform- and application-specific bioinformatics-based considerations. (Mardis, 2008).
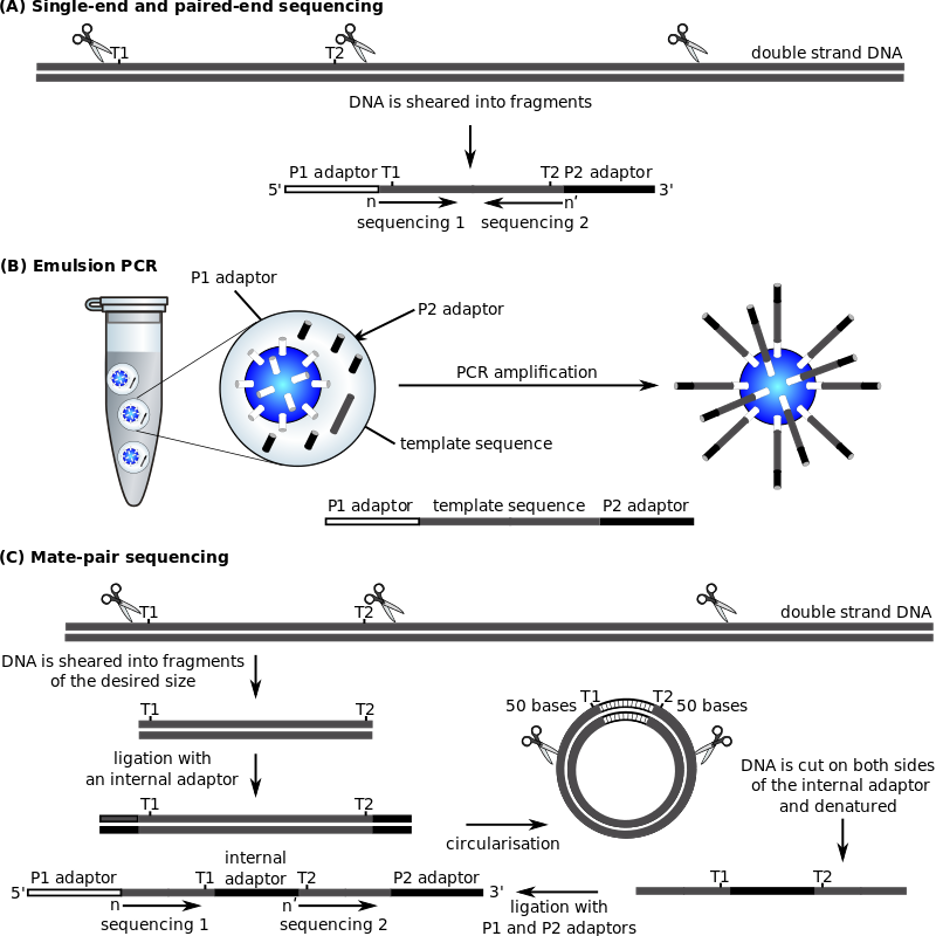
Figure 9.1: Steps of Applied Biosystems SOLiDTM Sequencer. (“ABI Solid Sequencing,” 2022).
DNA sequencing with 2D materials: in theory Modeling meets experimentation
Graphene Nanopores show promising potential for Mode I DNA sequencing applications. However, it is difficult to resolve individual bases because DNA passes too quickly through 2D nanoscale devices (including Nanopores, nanoribbons, and Nano gaps). Before we can actually use 2D materials for DNA sequencing, there are still some issues that need to be resolved. The success of DNA reading mechanisms that rely solely on nucleotide structure is questionable. A plausible pathway is a combination of different DNA reading mechanisms in DNA translocation through 2D materials. With advances in nanotechnology and theoretical modeling, we can expect optimal throughput toward nanoscale sequencers in the near future. (Liang et al., 2017)

Graphene Nanodevices for DNA sequencing
DNA sequencing could be one of the most disruptive innovations of the decade as it paves the way for personalized medicine. Due to its unique structure and properties, graphene offers opportunities to develop new sequencing technologies. We describe different approaches to using graphene nanodevices for DNA sequencing. In DNA sequencing, DNA is guided through Nanopores, Nano gaps, and nanoribbons. Graphene, a monolayer of carbon atoms arranged in a two-dimensional hexagonal lattice, offers new possibilities. Many different concepts have been proposed to sequence DNA using the unique properties of graphene. The atomically thin, ion-tight structure of graphene is the ultimate membrane for DNA sequencing. (Heerema & Dekker, 2016)
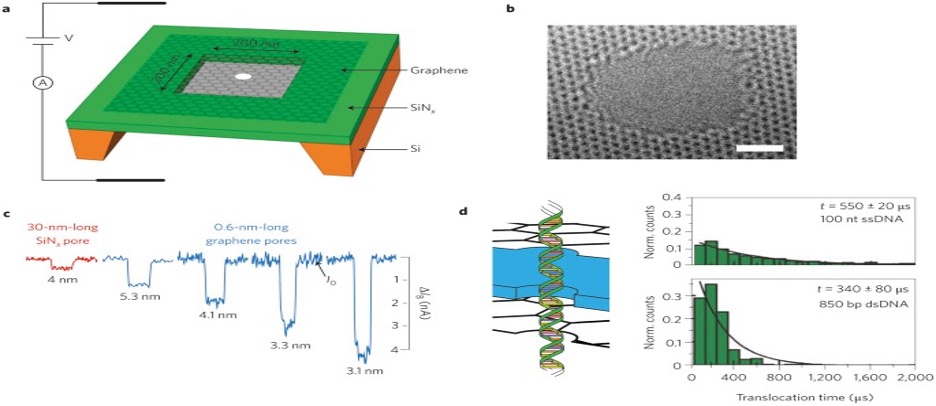
Figure 11.1: DNA detection with ionic current measurements through graphene Nano pores. (Heerema & Dekker, 2016)
Clinical applications of next-generation sequencing
Clinical applications of NGS testing involve several aspects of work. The first step is to decide which genetic mutations need to be investigated. Identification of possible mutations requires a review of the literature. It is also beneficial for laboratories to communicate with clinicians for each subspecialty. The next step is to establish a standard operating procedure (SOP). This includes creating SOPs and conducting preliminary tests to fine-tune the SOPs. After choosing an NGS method, you need to decide what kind of sample to test. Panels may be designed for both fresh and formalin-fixed paraffin-embedded (FFPE) tissue. After the NGS method is established, the assay should be validated by a CLIA-accredited lab. Validation plans typically define the positive and negative control samples to be used. Larger deletes and inserts make it harder for NGS to query them. AMP has issued guidelines for such a verification process. It aims to detect true variants and exclude false “variants” due to a noisy background. The validation process assesses analytical sensitivity, specificity, accuracy, precision, the limit of detection, sequencing depth, and cutoff allele frequency. Analytical sensitivity is a parameter that reflects the proportion of positive samples identified as true positives by a validated assay. Samples should be tested by at least two different technicians to assess reproducibility. Data are analyzed through a dedicated bioinformatics pipeline. Data analysis results are used to define the reference range and detection limit for the report. A quality control matrix ensures the quality of test results. (Qin, 2019)
Next-generation sequencing strategies
Twenty years after the first draft of the human genome sequence, developments and innovations in the field of genomics continue unabated. Research designs are increasing in scale for population-scale sequencings, such as the All of Us Research Project, while increasing resolution, such as the development and refinement of single-cell sequencing. The availability of low-cost, high-performance sequencing continues to expand the range of applications in genomics. Also, the development and revision of sequencing platforms, especially in long-read technology, have broadened the horizons of the nature and complexity of genome architecture. Population-scale projects reflect the changing challenges and opportunities of team-based science in terms of sample availability, sequencing and data-sharing techniques, and funding resources. The efficiency and impact of these and many other consortium projects on basic and translational research are immense. It is now unthinkable to perform any type of genomic analysis without using data from one or more consortium-based projects. Be it reference genomes, variant frequencies, sequence searches, or any other data type available in the public domain. (Levy & Boone, 2019).
GENERAL PRINCIPLES OF NEXT-GENERATION SEQUENCING
Next-generation sequencing tools do not require a cloning step per se. Instead, the DNA to be sequenced is used to construct a library of fragments with synthetic DNA covalently attached to each fragment end using DNA ligase. Each amplified fragment produces a single focus (a bead- or surface-supported cluster). Next-generation sequencing is often referred to as massively parallel, an apt description of the process that follows fragment amplification. Amplification is required to provide a sufficient signal from each DNA sequencing reaction step. These steps are performed in a format that allows hundreds of thousands to billions of reaction sites to be sequenced during each instrument run. Read length is the main difference between Sanger sequencing and next-generation sequencing data. With Sanger, read length is primarily determined by a combination of gel-related factors, including B. Percentage of polyacrylamide, separation time, gel length, and thickness. Read length is also a function of the signal-to-noise ratio. Noise sources vary by technology. (Mardis, 2013)
FUTURE POSSIBILITIES
DNA sequencing is becoming a more universal readout for an increasingly diverse set of front-end assays. Genome resequencing may be used to characterize strains or isolates relative to high-quality reference genomes such as C. elegans, Drosophila, and humans. Epigenomic variation has also been studied using next-generation sequencing approaches that allow the determination of methylation patterns across the genome.
References:
- ABI Solid Sequencing. (2022). In Wikipedia. https://en.wikipedia.org/w/index.php?title=ABI_Solid_Sequencing&oldid=1123467466
- Aly, S., & Bolbol, D. (2015). Next generation sequencing (NGS): A golden tool in forensic toolkit. Archives of Forensic Medicine and Criminology, 4, 260–271. https://doi.org/10.5114/amsik.2015.61029
- cdadmin. (n.d.). Sanger Sequencing: Introduction, Principle, and Protocol | CD Genomics Blog. Retrieved December 1, 2022, from https://www.cd-genomics.com/blog/sanger-sequencing-introduction-principle-and-protocol/
- DNA Sequencing | Springer Nature Experiments. (n.d.). Retrieved December 1, 2022, from https://experiments.springernature.com/articles/10.1007/978-1-0716-0334-5_44
- Figure 4. Illumina Genome Analyzer sequencing, the first of the Illumina platforms to be commercially launched. (2016). [Text]. American Society for Microbiology. https://www.ncbi.nlm.nih.gov/books/NBK513764/figure/fig4/
- Hall, N. (2007). Advanced sequencing technologies and their wider impact in microbiology. The Journal of Experimental Biology, 210, 1518–1525. https://doi.org/10.1242/jeb.001370
- Heerema, S. J., & Dekker, C. (2016). Graphene nanodevices for DNA sequencing. Nature Nanotechnology, 11(2), Article 2. https://doi.org/10.1038/nnano.2015.307
- Levy, S. E., & Boone, B. E. (2019). Next-Generation Sequencing Strategies. Cold Spring Harbor Perspectives in Medicine, 9(7), a025791. https://doi.org/10.1101/cshperspect.a025791
- Liang, L., Shen, J.-W., Zhang, Z., & Wang, Q. (2017). DNA sequencing by two-dimensional materials: As theoretical modeling meets experiments. Biosensors and Bioelectronics, 89, 280–292.https://doi.org/10.1016/j.bios.2015.12.037
- Mardis, E. R. (2008). Next-generation DNA sequencing methods. Annual Review of Genomics and Human Genetics, 9, 387–402. https://doi.org/10.1146/annurev.genom.9.081307.164359
- Mardis, E. R. (2013). Next-generation sequencing platforms. Annual Review of Analytical Chemistry (Palo Alto, Calif.), 6, 287–303. https://doi.org/10.1146/annurev-anchem-062012-092628
- Porreca, G. J., Shendure, J., & Church, G. M. (2006). Polony DNA Sequencing. Current Protocols in Molecular Biology, 76(1), 7.8.1-7.8.22. https://doi.org/10.1002/0471142727.mb0708s76
- Qin, D. (2019). Next-generation sequencing and its clinical application. Cancer Biology & Medicine, 16(1), 4–10. https://doi.org/10.20892/j.issn.2095-3941.2018.0055
- Rizzo, J. M., & Buck, M. J. (2012). Key Principles and Clinical Applications of “Next-Generation” DNA Sequencing. Cancer Prevention Research, 5(7), 887–900. https://doi.org/10.1158/1940-6207.CAPR-11-0432
- Shotgun Sequencing—An overview | ScienceDirect Topics. (n.d.). Retrieved December 1, 2022, from https://www.sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/shotgun-sequencing
- Voelkerding, K., Dames, S., & Durtschi, J. (2009). Next-Generation Sequencing: From Basic Research to Diagnostics. Clinical Chemistry, 55, 641–658. https://doi.org/10.1373/clinchem.2008.112789
If you like this article, you can go through our other top articles
- Microneedles: An Intelligent Technology for Transdermal Drug Delivery – https://learnlifescience.com/microneedles-an-intelligent-technology-for-transdermal-drug-delivery/
- Regenerative food production: A sustainable food production method – https://learnlifescience.com/regenerative-food-production-a-sustainable-food-production-method/
- BIOFILM AND ITS ROLE IN BIOREMEDIATION – https://learnlifescience.com/biofilm-and-its-role-in-bioremediation/
“Since the article has been written to reflect the actual views and capabilities of the author(s), they are not revised for content and only lightly edited to be confirmed with the Learn life sciences style guidelines”






Reading your article helped me a lot and I agree with you. But I still have some doubts, can you clarify for me? I’ll keep an eye out for your answers.
Cheers
Thanks for sharing. I read many of your blog posts, cool, your blog is very good.